Каталог статей
| Главная » Статьи » Объявления |
Болезнь Вильсона-Коновалова
Это означает, что заболевание является наследственным и передается ребенку от родителей в случае наличия у обоих родителей патологического гена. При болезни Вильсона-Коновалова за возникновение отвечает мутированный ген ATP7B в 13 парной хромосоме. При болезни Вильсона-Коновалова в организме нарушается обмен меди, что приводит к накоплению ее избыточного количества и снижения процессов ее вывода. Для данной патологии характерно сочетанием дистрофических изменений в головном мозге и поражения печени – циррозом . Поэтому выделяют следущие формы болезни Вильсона-Коновалова:
Начало заболевания при печеночной форме проявляется обычно в юношеском возрасте, примерно с 10-15 лет. Поражение центральной нервной системы при болезни Вильсона-Коновалова имеет более длительный срок развития и обнаруживается у пациентов старше 20 лет. Носителями мутированного гена является приблизительно 0,6% мирового населения, при этом заболевание встречается в 3 случаях на 100 000 жителей. Прогноз болезни Вильсона-Коновалова значительно выше при рождении ребенка в родственном браке. В таких случаях существует реальная угроза наследования мутированного гена от обоих родителей. Симптомы болезни Вильсона-КоноваловаВ формировании заболевания отмечаются две стадии – скрытую и клиническую, когда появляются признаки разрушения. Печеночная форма болезни проявляется повышением уровня ферментов – аминотрансферазом и признаки ее обнаруживаются обычно к 7 годам. Избыточное скопление меди в организме приводит к острому началу болезни. При этом симптомы болезни Вильсона-Коновалова выражаются в резком повышении температуры, желтушной окраске кожи, развитии астенического синдрома. Прогрессирование болезни приводит к накоплению жировых масс в печени и печеночной недостаточности. Симптомы болезни Вильсона-Коновалова, обусловленной поражением центральной нервной системы, проявляются в расстройствах речевой функции, ослаблении мимики, повышенном слюноотделении, треморе и сложностях в координации движений. При этом у пациентов не наблюдается снижение интеллектуальных способностей, но поведение может быть непредсказуемым – от агрессии до подавленности. Возможно появление различных фобий. Прогноз болезни Вильсона-Коновалова без соответствующего лечения неутешителен, т.к. при этом, наряду с перечисленными патологиями, нарушается деятельность сердца, почек, поражаются суставы и кости. Завершающая стадия болезни приводит к коматозному состоянию. Продолжительности жизни при заболевании без необходимого систематического и пожизненного лечения составляет от 5 до 15 лет. Диагностика болезни Вильсона-Коновалова
Далее проводится лабораторное обследование пациента – исследуется состояние мочи и крови, определяется уровень печеночных ферментов и выделение меди в суточной урине. Диагностика болезни Вильсона-Коновалова включает инструментальные методы обследования - проводится компьютерная и магниторезонансная томография, ультразвуковое исследование. Этими методами определяются гепато и спленгомегалия, т.е. устанавливаются степень роста печени и селезенки. Устанавливается фаза разрушения в головном мозге подкорковых нейронных узлов. Для выявления мутированного гена при генетической диагностике болезни Вильсона-Коновалова проводится тестирование крови больного и его близких родственников. По результатам диагностирования назначается корректировка пораженных органов. Лечение болезни Вильсона - КоноваловаТерапия заболевания носит симптоматический характер. Основной целью при лечении болезни Вильсона-Коновалова является снижение объема поступающей и уже имеющейся в организме меди. Для вывода избытков находящихся в составе организма меди используются специальные препараты – соли цинка и D-пеницилламин. Консервативное лечение болезни Вильсона-Коновалова с помощью медикаментов ведется строго по индивидуальной схеме с постепенным увеличением дозировки. Проводится гепатопротекторная терапия, способствующая улучшению деятельности печени. При диагностировании заболевания помимо приема препаратов необходимо проходить регулярные профилактические обследования не реже одного раза в год, но все, же желательно проходить осмотр каждые 6 месяцев. При этом обязателен контроль над состоянием мочи и крови, а также определяется состояние функций почек и печени. В случае развития печеночной недостаточности рекомендована трансплантация донорской здоровой печени. Такой метод, при приживаемости пересаженного органа, способствует полному выздоровлению, но только от цирроза, а не от болезни в целом. Кроме того при болезни Коновалова-Вильсона диета занимает одно из важнейших мест в успешном лечении. Для этого в питании необходимо избегать продуктов с высоким содержанием меди – шоколад, орехи, бобовые, кофе и т.д. Диета при болезни Коновалова-Вильсона включает комплекс мер для похудения и подразумевает постоянное ее использование в течение жизни. Кроме того алкоголь и минеральная вода полностью исключается из употребления. В диету при болезни Коновалова-Вильсона не включаются печень, почки, картофель, баранина, свинина, ржаной хлеб. Для приготовления пищи медную посуду использовать нельзя. В связи с тем, что прогноз болезни Вильсона-Коновалова не всегда благоприятен, а причины его развития имеют наследственные факторы, единственным профилактическим мероприятием является генетическая консультация для родителей, как уже имеющих ребенка с данным заболеванием, так и тех, которые собираются стать родителями в родственном браке. Описание:Болезнь Вильсона-Коновалова (гепатолентикулярная дегенерация)nbspnbsp - редкое наследственное заболевание, в основе которого лежит нарушение метаболизма меди и накопление ее в печени и других внутренних органах. Заболевание протекает с преимущественным поражением печени и ЦНС, вовлечением в патологический процесс органа зрения и почек. Симптомы Болезни Вильсона-Коновалова:Выделяют три стадии болезни: nbspnbsp nbspnbsp *I - латентную с неспецифическими морфологическими изменениями печени или мелко- или среднекапельный жировой дистрофией nbspnbsp nbspnbsp *II - ХАГ nbspnbsp nbspnbsp *III - цирротическую стадию. Возможно длительное субклиническое течение, при котором в пунктатах печени находят некрозы единичных гепатоцитов, жировую инфильтрацию, перипортальный фиброз. У больных с клинически латентным течением диагноз ставят при выявлении характерных изменений показателей метаболизма меди: nbspnbsp 1.снижения уровня церулоплазмина плазмы ( 100 мкг/сут.) экскреции меди с мочой. Патогномонично наличие роговичного кольца Кайзера-Флейшера шириной около 2 мм вследствие накопления серовато-коричневого с зеленоватым оттенком содержащего медь пигмента кнутри от лимба на задней поверхности роговицы. Оно лучше всего выявляется с помощью щелевой лампы. У большинства больных определяют гепатомегалию и более чем у 50% - спленомегалию. 5% больных моложе 35 лет с картиной ХАГ неясной этиологии страдают болезнью Вильсона-Коновалова. Поэтому у всех молодых больных ХАГ целесообразно определение показателей обмена меди. Гепатит может протекать с астенизацией, желтухой, болью в животе и диспепсическими явлениями, гипераминотрансфераземией, гипоальбуминемией и умеренной гипергаммаглобулинемией. Часто наблюдают удлинение протромбинового времени, геморрагический диатез. гемолиз. Возможен костно-суставной синдром с остеопорозом или остеомаляцией, поражением коленных суставов и позвоночника. Поражение почек проявляется периферическими отеками, микрогематурией, незначительной протеинурией, увеличением концентрации мочевины и креатинина крови. Часто наблюдается нарушение реабсорбции на уровне проксимальных канальцев и в связи с этим гипераминоацидурия, гиперкальциурия. гиперфосфатурия, нарастание экскреции с мочой мочевой кислоты и натрия. Возможно поражение дистальных канальцев с развитием почечного канальцевого ацидоза. Описаны больные, у которых болезнь Вильсона-Коновалова дебютировала картиной острого гепатита или фульминантной печеночной недостаточности с явлениями гемолиза, развившейся после гриппоподобного эпизода или периода диспепсических нарушений. Начальным проявлением болезни может быть также острая почечная недостаточность. обусловленная массивным гемолизом. Конечная стадия болезни Вильсона-Коновалова - стадия сформированного цирроза печени. чаще макронодулярного, с накоплением меди в перипортальных пространствах и вдоль фиброзных септ. Цирроз часто сопровождается гиперспленизмом с тромбоцитопенией и геморрагическим синдромом. Ранние неврологические проявления болезни у взрослых - тремор и дизартрия. напоминающая скандированную речь, непроизвольные резкие повороты головы и взгляда в ту же сторону, нарушения координации движений, эсобенно заметные при ходьбе, дисфагия. дистония. Причины Болезни Вильсона-Коновалова:И.А. Иванова-Смоленская профеcсор, доктор медицинских наук ГУ НИИ неврологии РАМН Болезнь Вильсона–Коновалова (гепато-лентикулярная дегенерация) относится к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов. Если своевременно не начать лечение, направленное на выведение токсичных избытков меди из организма, то через 5–7 лет больной обречен на смерть. Болезнь поражает 25% братьев и сестер в семье при клинически здоровых родителях, которые являются носителями аномального гена (аутосомно-рецессивный тип наследования). Заболевают только те индивидуумы, которые унаследовали два мутантных гена, то есть по одному от матери и от отца – гомозиготные носители мутации лица, которые от одного из родителей получили мутантный ген, а от другого – нормальный ген, являются гетерозиготными носителями мутации и остаются здоровыми. Открытый недавно ген болезни отвечает за синтез медь-транспортирующего белка (АТР7В). При гепатолентикулярной дегенерации обмен меди и медьсодержащих белков нарушается, появляется избыток “свободной” меди, которая в больших количествах откладывается в печени, мозге, роговице, а также выделяется с мочой. Не случайно диагностика болезни базируется на обнаружении характерных нарушений медного обмена. Благодаря идентификации гена в настоящее время возможна и ДНК- диагностика этого заболевания. Поражение печени избытком “свободной” меди проявляется циррозом печени. Поражение мозга приводит к развитию тяжелой неврологической симптоматики: дрожанию конечностей и всего туловища, повышению мышечного тонуса, иногда сопровождающемуся болезненными спазмами, нарушением речи, глотания, снижению интеллекта. Отложение меди в роговице (по краю радужной оболочки) обусловливает формирование кольца Кайзера–Флейшера – буро-зеленоватого пигмента. По этому признаку диагноз болезни можно поставить безошибочно. Гепато-лентикулярная дегенерация известна с глубокой древности. Дошедшее до нас изображение египетского фараона Тутанхамона, по мнению крупнейшего специалиста J. Walshe, не исключает вероятности, что он страдал этим заболеванием. Институт неврологии РАМН в течение многих лет занимается проблемой гепато-лентикуляной дегенерации. Знаменитый отечественный невролог академик АМН СССР Н.В. Коновалов, один из основателей Института, посвятил этому заболеванию две монографии, последняя из которых в 1964 году была удостоена Ленинской премии. В последующие годы данное заболевание продолжало успешно изучаться сотрудниками нейрогенетического отделения Института неврологии, под наблюдением которых за 40 лет находилось свыше500 семей, отягощенных этим недугом. Весь многолетний опыт Института свидетельствует о том, что ключевой проблемой является ранняя диагностика гепатолентикулярной дегенерации. Чем раньше начать лечение (в идеале – еще на досимптоматической стадии либо на доневрологическом этапе, то есть до появления признаков поражения мозга), тем лучше эффект. Вот почему, если в семье есть хоть один ребенок, страдающий этим заболеванием, необходимо тщательное обследование всех его братьев и сестер, в том числе с использованием самых современных биохимических и молекулярно-генетических методов. Заподозрить раннюю стадию болезни можно на основании следующих признаков: перенесенной желтухи повторных кровотечений из носа, кровоточивости десен либо множественных кровоподтеков сосудистых “звездочек” на коже груди и спины своеобразных “полосок” (белых, меняющих периодически окраску на красновато-синюшную) на бедрах и в подмышечных областях гормональных нарушений в виде аменореи или дисменореи у девушек, гинекомастии (нагрубание грудных сосков) у юношей, а также акромегалии(увеличение носа, подбородка, утолщение губ) снижения интеллекта и изменений психики в виде чередования дурашливости и пониженного настроения, трудностей усвоения нового материала, проблем с успеваемостью в школе. Гепато-лентикулярная дегенерация может начать проявляться в детском, подростковом, юношеском, зрелом возрасте и очень редко – в 50–60 лет. Чем раньше начинается заболевание, тем тяжелее оно протекает (при отсутствии лечения). Однако болезнь Вильсона–Коновалова – редкий пример наследственного нарушения, для которого разработаны высокоэффективные методы лечения: даже при появлении тяжелой неврологической симптоматики систематическое лечение обычно дает “драматический” эффект, вплоть до исчезновения всех симптомов или резкого их уменьшения. Пациенты вновь могут полностью обслуживать себя, вести домашнюю работу, учиться, работать по профессии, создать семью и родить здорового ребенка (под нашим наблюдением находятся 30 женщин, страдающих гепатолентикулярной дегенерацией и благополучно родивших здоровых детей). Пациентам с гепатолентикулярной дегенерацией необходимо регулярно наблюдаться у постоянного лечащего врача. В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин. Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности. При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). Нами было предложено комбинированное лечение D-пеницилламином и препаратами цинка, что дает возможность снизить дозу и избежать побочных явлений. У больных в пресимптоматической стадии достаточно лечения только препаратами цинка. В настоящее время за рубежом в тяжелых случаях болезни, не поддающихся консервативному лечению, широко применяется пересадка печени. При удачном исходе операции больной полностью выздоравливает и не нуждается в дальнейшем приеме препаратов. В России делаются первые шаги в этом направлении, и одним из таких шагов является разработанный нами совместно с Институтом трансплантологии и искусственных органов метод био-гемоперфузии с изолированными живыми клетками печени и селезенки – так называемый аппарат “вспомогательная печень”. Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени. Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики. © Журнал Нервы , 2006, №4 Источники: http://promedicinu.ru/diseases/boliezn-vilsona-konovalova, http://www.24farm.ru/narushenie_obmena_veshev/bolezn_vilsona_konovalova/, http://www.neurology.ru/patient/a-IvanovaSmolenskaya2006-4.htm | |
| Просмотров: 790 | |
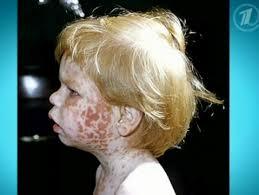 Тяжелое и редко встречающееся генетическое заболевание - гепатолентикулярная дегенерация или гепатоцеллюлярная дистрофия, иначе называемое болезнью Вильсона-Коновалова обусловлено аутоиммунно-рецессивном типом наследования.
Тяжелое и редко встречающееся генетическое заболевание - гепатолентикулярная дегенерация или гепатоцеллюлярная дистрофия, иначе называемое болезнью Вильсона-Коновалова обусловлено аутоиммунно-рецессивном типом наследования. При физикальном обследовании пациента подозрением на болезнь Вильсона-Коновалова может служить типичный признак - кольцо Кайзера-Флейшера, выражающийся коричнево-желтой обводкой по периферии глазной роговицы.
При физикальном обследовании пациента подозрением на болезнь Вильсона-Коновалова может служить типичный признак - кольцо Кайзера-Флейшера, выражающийся коричнево-желтой обводкой по периферии глазной роговицы.| Всего комментариев: 0 | |